Bacterial genotyping and phylogenetic analysis
Today we will be extracting the 16S rDNA sequences from our genome assemblies and using the sequences to help understand the evolutionary relationships between our isolates.
First, change directories into the folder containing your genome assembly:
cd ~/genome_assembly/kmer75assembly/
You will need to download the reference 16S rDNA sequence from Agrobacterium fabrum C58 using this command:
cp /nfs1/Teaching/home/weisbeal_ws/bin/C58_16S.fna ./
Next, we will use the genomic sequence that we generated yesterday using velvet as the subject in a BLAST search with the C58 sequence as a query. You will use a script to automate the following functions:
1. Generate BLASTDB from genomic sequence
2. Use BLASTN+ to find the 16S rDNA in your genomic sequence
3. Extract the 16S rDNA sequence from the BLAST results and save it to a file
Run the command:
/nfs1/Teaching/home/weisbeal_ws/bin/16S_blast.sh contigs.fa
The output you are looking for is called 16S.fasta. You can make sure your file was generated using the ls command, and you can view your sequence by using the cat 16S.fasta command. This will print the sequence to your screen.
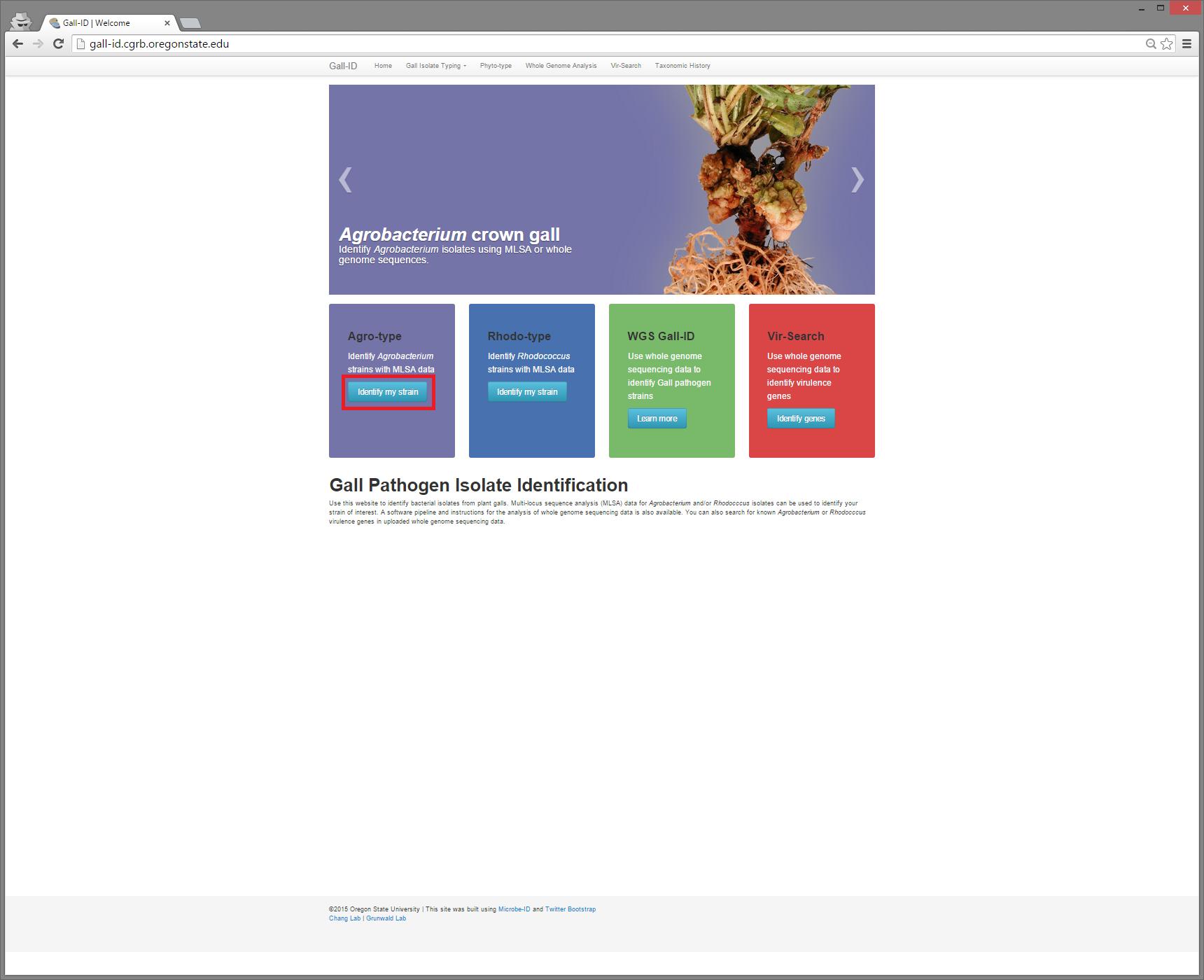
Next, we will be using the Gall-ID website to generate a phylogenetic tree including your 16S sequences. First, go to http://gall-id.cgrb.oregonstate.edu/ and click on Identify my strain under Agro-type (See red boxes for hints!)
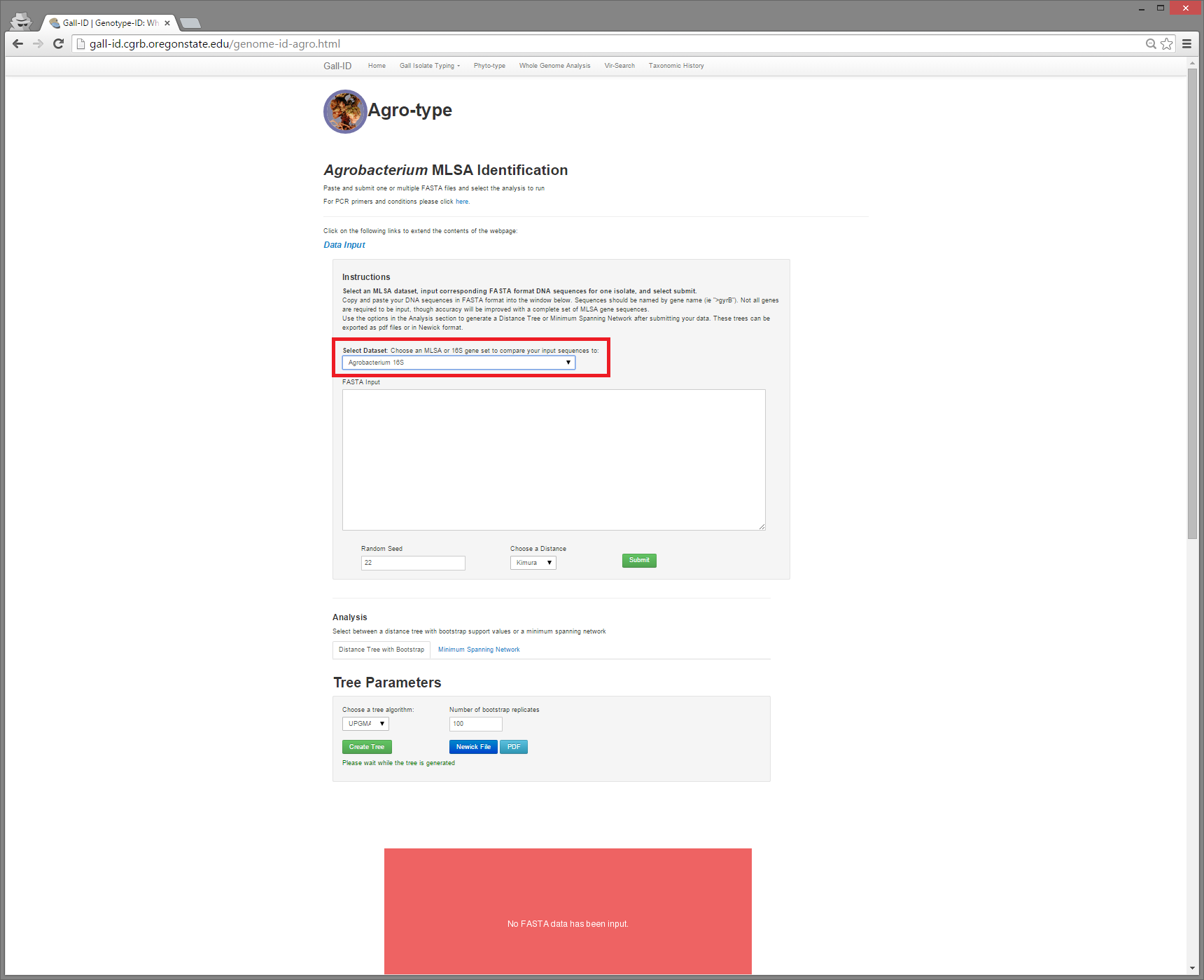
Next, choose Agrobacterium 16S on the Select Dataset drop-down menu
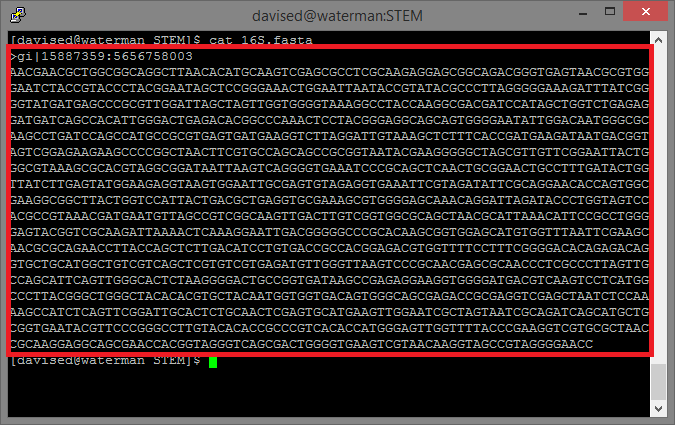
In your putty window, run the command:
cat 16S.fasta
Copy your fasta sequence from your putty window by highlighting the text, including the header line, which starts with ">". By highlighting the text, your sequence will be automatically copied to your clipboard.
Paste your sequence into the text box on the gall-id website, and change the header line to read >16S
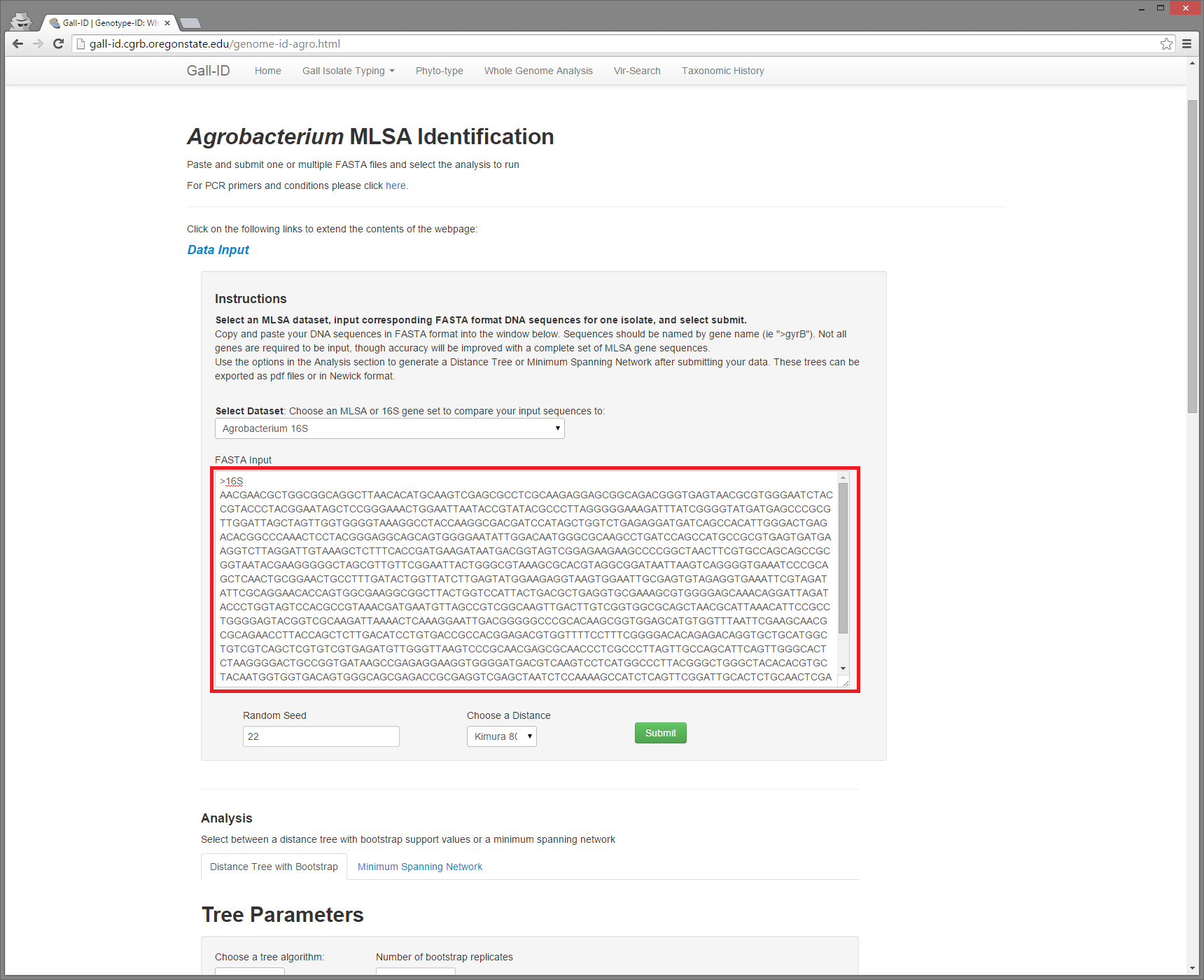
Click on the green submit button and wait for your tree to be generated! Your sequence will be labeled as query isolate (Results not shown!)
While you are waiting for the tree to generate, we will download your 16S sequence and combine it with everyone else’s 16S sequences and provide the file to you.
Once it is done, you can download it here:
http://dnacamp.cgrb.oregonstate.edu/16S_combined.fasta
You can examine your trees to see the closest relatives with sequences found in the NCBI nucleotide database.
Next, we will align our sequences and generate a tree to see how our isolates are related to one another.
Open MEGA 6 from your desktop
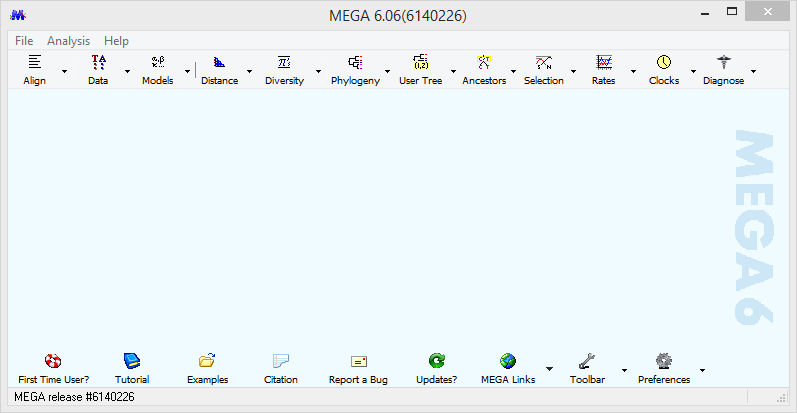
Click on Align -> Edit/Build alignment and then Retrieve sequences from File and OK. Navigate to the location of 16S_concat.fasta and click the file name, then click Open.
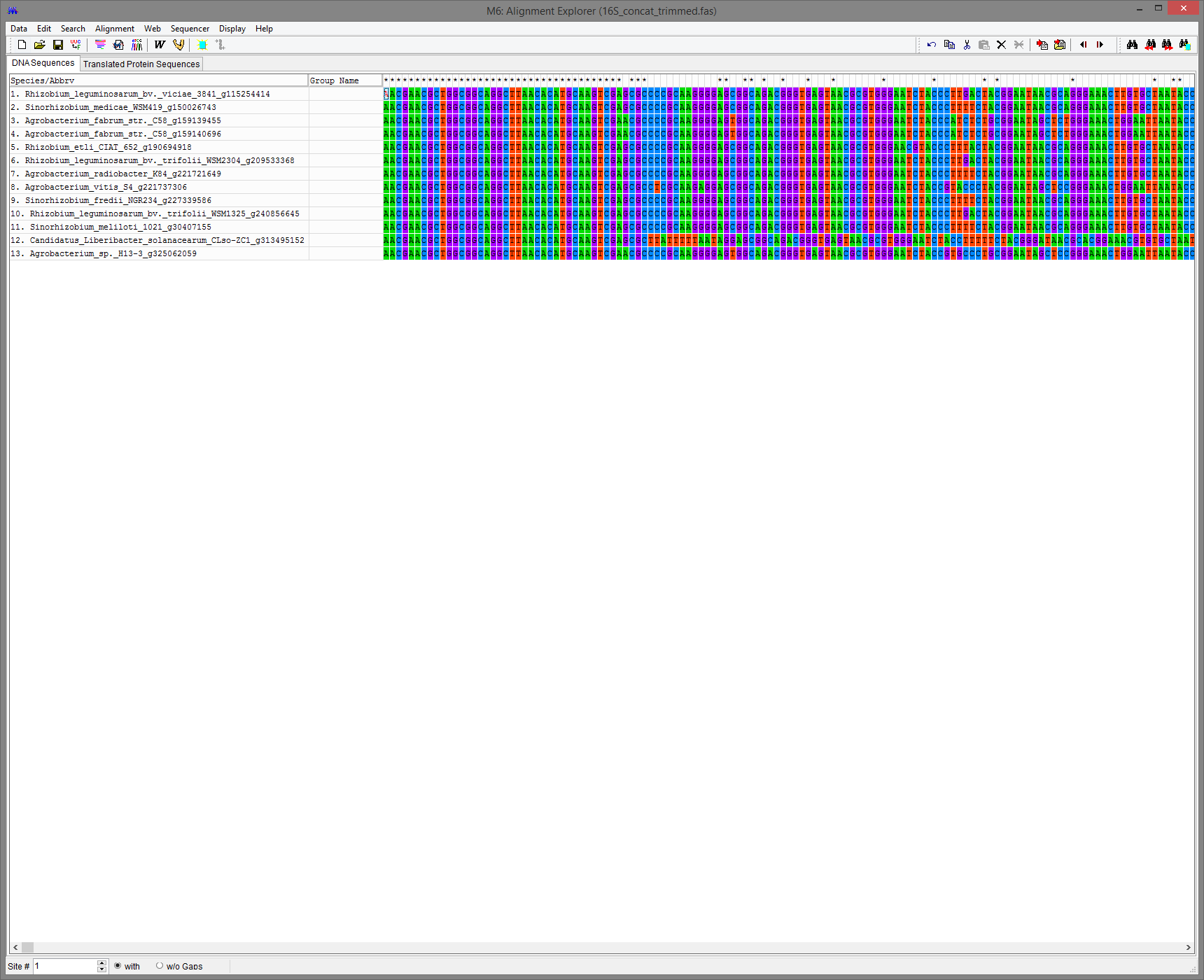
Click on the Muscle tool to align the sequences using MUSCLE (Align DNA, not codons, and click OK when MEGA asks to select all)
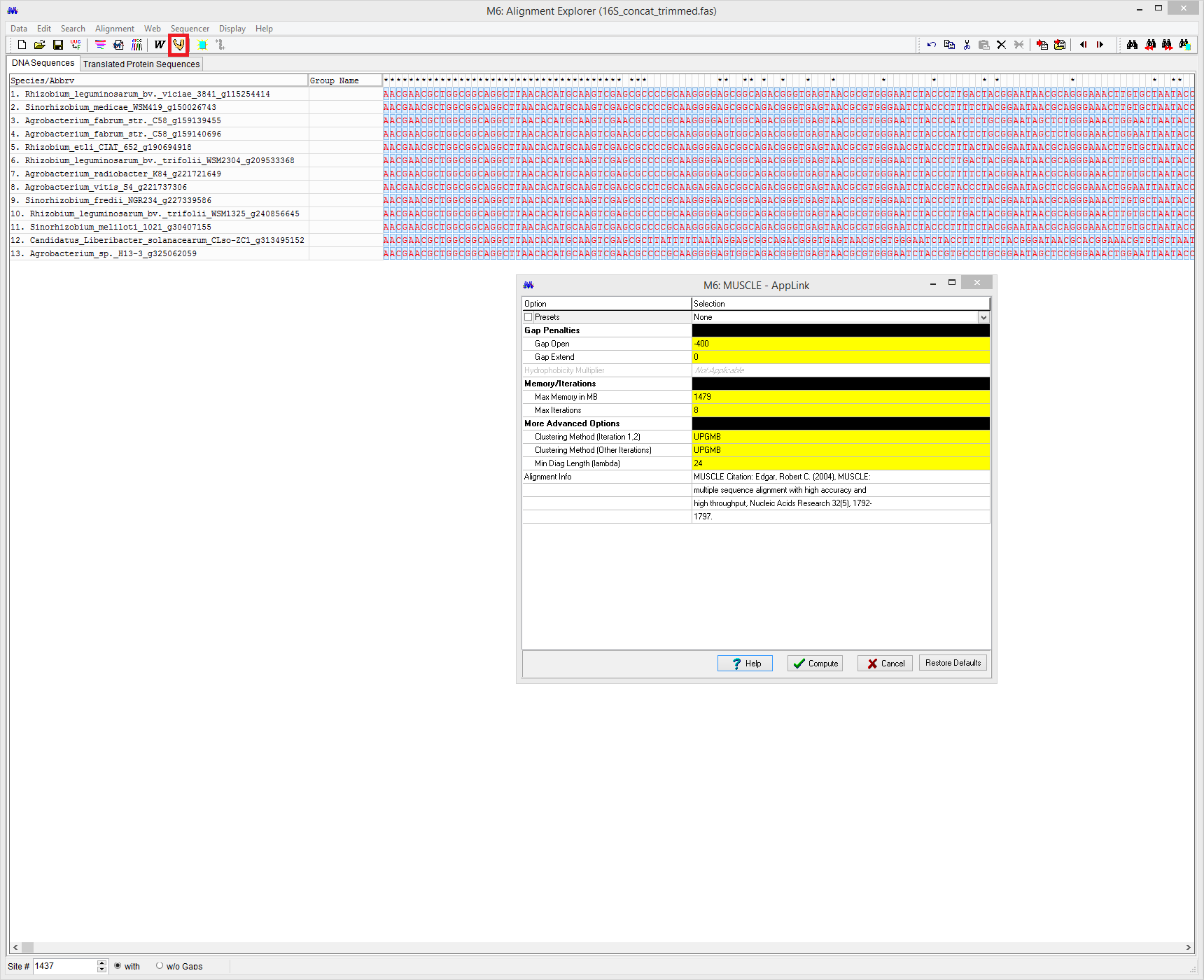
Default values are fine. Click compute to generate a multiple sequence alignment. Once the sequences are aligned, click Data then select Phylogenetic analysis from the drop-down menu. Select NO for protein-coding nucleotide sequence data. Click on the original MEGA window. You should see a TA and Close Data button indicating that the data has been opened to phylogenetic analysis.
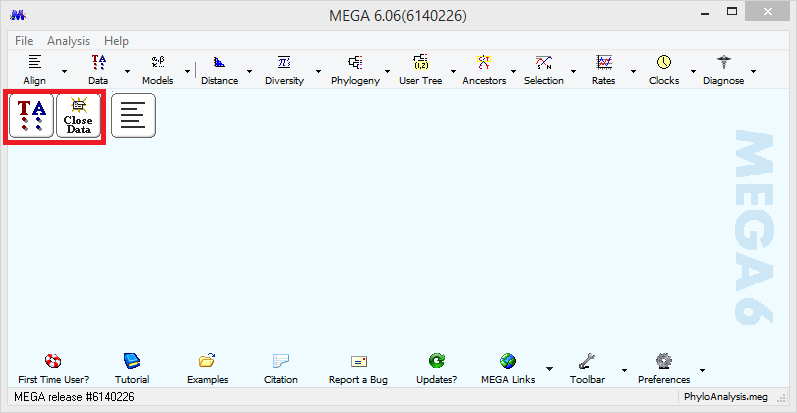
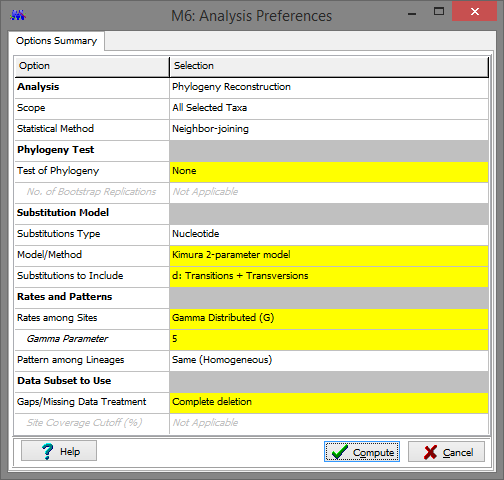
Next, click Phylogeny, Construct/Test Neighbor-Joining Tree, and Yes to use the currently active data. Change the Model/Method to Kimura 2-parameter model to keep things consistent between the Gall-ID site and MEGA, then click Compute.
You will then have your tree and can compare/contrast with the tree generated using Gall-ID.
First, change directories into the folder containing your genome assembly:
cd ~/genome_assembly/kmer75assembly/
You will need to download the reference 16S rDNA sequence from Agrobacterium fabrum C58 using this command:
cp /nfs1/Teaching/home/weisbeal_ws/bin/C58_16S.fna ./
Next, we will use the genomic sequence that we generated yesterday using velvet as the subject in a BLAST search with the C58 sequence as a query. You will use a script to automate the following functions:
1. Generate BLASTDB from genomic sequence
2. Use BLASTN+ to find the 16S rDNA in your genomic sequence
3. Extract the 16S rDNA sequence from the BLAST results and save it to a file
Run the command:
/nfs1/Teaching/home/weisbeal_ws/bin/16S_blast.sh contigs.fa
The output you are looking for is called 16S.fasta. You can make sure your file was generated using the ls command, and you can view your sequence by using the cat 16S.fasta command. This will print the sequence to your screen.
Next, we will be using the Gall-ID website to generate a phylogenetic tree including your 16S sequences. First, go to http://gall-id.cgrb.oregonstate.edu/ and click on Identify my strain under Agro-type (See red boxes for hints!)
Next, choose Agrobacterium 16S on the Select Dataset drop-down menu
In your putty window, run the command:
cat 16S.fasta
Copy your fasta sequence from your putty window by highlighting the text, including the header line, which starts with ">". By highlighting the text, your sequence will be automatically copied to your clipboard.
Paste your sequence into the text box on the gall-id website, and change the header line to read >16S
Click on the green submit button and wait for your tree to be generated! Your sequence will be labeled as query isolate (Results not shown!)
While you are waiting for the tree to generate, we will download your 16S sequence and combine it with everyone else’s 16S sequences and provide the file to you.
Once it is done, you can download it here:
http://dnacamp.cgrb.oregonstate.edu/16S_combined.fasta
You can examine your trees to see the closest relatives with sequences found in the NCBI nucleotide database.
Next, we will align our sequences and generate a tree to see how our isolates are related to one another.
Open MEGA 6 from your desktop
Click on Align -> Edit/Build alignment and then Retrieve sequences from File and OK. Navigate to the location of 16S_concat.fasta and click the file name, then click Open.
Click on the Muscle tool to align the sequences using MUSCLE (Align DNA, not codons, and click OK when MEGA asks to select all)
Default values are fine. Click compute to generate a multiple sequence alignment. Once the sequences are aligned, click Data then select Phylogenetic analysis from the drop-down menu. Select NO for protein-coding nucleotide sequence data. Click on the original MEGA window. You should see a TA and Close Data button indicating that the data has been opened to phylogenetic analysis.
Next, click Phylogeny, Construct/Test Neighbor-Joining Tree, and Yes to use the currently active data. Change the Model/Method to Kimura 2-parameter model to keep things consistent between the Gall-ID site and MEGA, then click Compute.
You will then have your tree and can compare/contrast with the tree generated using Gall-ID.