Bioinformatics: Assessing Read Quality
In this project we will be evaluating the read quality of the DNA sequences you sequenced yesterday.
DNA sequencing is a process were individual DNA fragments are "sequenced", meaning the individual nucleotides of A, C, G, and T are determined for each specific DNA fragment. In addition to the DNA nucleotides, the sequencing process produces quality scores, also encoded by characters. There is one quality character for every DNA nucleotide character. Each of these "quality characters" corresponds to the probability that the DNA nucleotide is an error.
Let's look at an example fastq file:
The quality characters are encoded as ascii characters. First let's see how the quality scores PHRED scores are defined in terms of "P", the probability of an error:
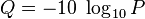
So the smaller the error probability, the higher the quality. That is, the less likely the sequenced nucleotide is an error, the better the probability.
To go from a quality character to a quality score, we can use the python function "ord". This will give us the ascii number. Here is a quick example for the letter A:
Here is a full ascii table with numbers: ASCII table
The truth is, to go from quality character to PHRED score, you need:
Qnumber = ord(Qcharacter) - 33
However python has tools that can read in a fastq file and give you quality scores. To test this, let's create a short version of the fastq file. We can use the command "head" to print some number of lines of a file:
head -4 agro_reads/Atumefaciens_####_R1.fastq > test.fastq
Now we can test this one out. You may want to print the file to the screen:
cat test.fastq
So this should have 4 lines. Does it? Now let's enter the python shell:
python
The first thing we want to do is import tools to read in a fastq file:
from Bio import SeqIO
Now let's read in the fastq file. This command will process the lines appropriately. We need to give it the file name, and the type of file ("fastq"):
data = SeqIO.parse("test.fastq","fastq")
Now let's try and print out the PHRED scores. The hard part here is that we need to indent the second and third lines for python to properly interpret the "for loop".Note: the best way to indent these lines is by hitting "tab" once the second line, and twice for the third line. Try this, but be aware this will print a lot of stuff to the screen (the PHRED scores):
Now we know that we are getting quality scores. I have a script you can download with this command:
wget http://dnacamp.cgrb.oregonstate.edu/printAverageQualityScores.py
And let's move it to your scripts directory:
mv printAverageQualityScores.py ~/scripts/.
Let's look at the script in detail:
wget http://dnacamp.cgrb.oregonstate.edu/untrimmed_reads.fastq
Finally, we can run the script like this:
python ~/scripts/printAverageQualityScores.py untrimmed_reads.fastq
Transfer the file with Filezilla. How does it look?
DNA sequencing is a process were individual DNA fragments are "sequenced", meaning the individual nucleotides of A, C, G, and T are determined for each specific DNA fragment. In addition to the DNA nucleotides, the sequencing process produces quality scores, also encoded by characters. There is one quality character for every DNA nucleotide character. Each of these "quality characters" corresponds to the probability that the DNA nucleotide is an error.
Let's look at an example fastq file:
@DB775P1:316:C4AGUACXX:3:1101:1223:1926 1:Y:0:AGTCAA NGCACTNGAGAAGCTCCGACTGGAGNGCAACNCGGCCACCGTCTGTGGCNAGGNCTTTAAGAACGNGGANCCNNCCTACAGCTGCC + #0;@@@#2<@@?@@@?@@@@??@>@#18=??#0?;3<<#0059=?>?9>?7668>1#-;96;?1>8=??=;?##################### @DB775P1:316:C4AGUACXX:3:1101:1471:1942 1:N:0:AGTCAA NTTTATTTCTTTATTTTCATTAATTTTATTTNATAATTTCATAGGATTANTTCCATATATTTTTACAAGAACANGACATTTAACTT + #1=;DDEFHHGGFHIIIJGIJJJJJJJAHIJ#2AEGHIJIIJJJJEGI@#0?FFHHIIHIJJJJGIIJJJJJI#-5?EEHEFFFFFThe fastq file is composed of quartets of lines. The first is the ID, the second is the sequence itself, the third is "+" or optionally repeating the ID. The fourth line is the quality scores (quality characters)
The quality characters are encoded as ascii characters. First let's see how the quality scores PHRED scores are defined in terms of "P", the probability of an error:
So the smaller the error probability, the higher the quality. That is, the less likely the sequenced nucleotide is an error, the better the probability.
To go from a quality character to a quality score, we can use the python function "ord". This will give us the ascii number. Here is a quick example for the letter A:
$ python
Python 2.7.8 (default, Aug 7 2014, 09:56:10)
[GCC 4.4.7 20120313 (Red Hat 4.4.7-4)] on linux2
Type "help", "copyright", "credits" or "license" for more information.
>>> ord('A')
65
>>> quit()
Here is a full ascii table with numbers: ASCII table
{kind=link}
The truth is, to go from quality character to PHRED score, you need:
Qnumber = ord(Qcharacter) - 33
However python has tools that can read in a fastq file and give you quality scores. To test this, let's create a short version of the fastq file. We can use the command "head" to print some number of lines of a file:
head -4 agro_reads/Atumefaciens_####_R1.fastq > test.fastq
Now we can test this one out. You may want to print the file to the screen:
cat test.fastq
So this should have 4 lines. Does it? Now let's enter the python shell:
python
The first thing we want to do is import tools to read in a fastq file:
from Bio import SeqIO
Now let's read in the fastq file. This command will process the lines appropriately. We need to give it the file name, and the type of file ("fastq"):
data = SeqIO.parse("test.fastq","fastq")
Now let's try and print out the PHRED scores. The hard part here is that we need to indent the second and third lines for python to properly interpret the "for loop".Note: the best way to indent these lines is by hitting "tab" once the second line, and twice for the third line. Try this, but be aware this will print a lot of stuff to the screen (the PHRED scores):
for record in data:
for Q in record.letter_annotations["phred_quality"]:
print Q
Note: you'll need to hit "enter" again after all this!Now we know that we are getting quality scores. I have a script you can download with this command:
wget http://dnacamp.cgrb.oregonstate.edu/printAverageQualityScores.py
And let's move it to your scripts directory:
mv printAverageQualityScores.py ~/scripts/.
Let's look at the script in detail:
import sys
from Bio import SeqIO
import matplotlib as mpl
mpl.use('Agg')
import matplotlib.pyplot as pyplot
# this section takes care of reading in data from user
if len(sys.argv) != 2 or "-h" in sys.argv or "--help" in sys.argv:
print >> sys.stderr, "Usage: printAverageQualityScores.py "
sys.exit()
# read in the fastq file name
fastq = sys.argv[1]
# parse the fastq file
data = SeqIO.parse(fastq,"fastq")
# initialize a list of probabilities
pList = []
length = 0
for record in data:
pArray = []
for Q in record.letter_annotations["phred_quality"]:
# convert the PHRED score to a probability
p = 10**(-float(Q)/10)
# append this specific probabity to array for this sequence
pArray = pArray + [p]
# now append the sequence's probablity to a list of all of them
pList = pList + [pArray]
# store the length of the probability array (same as length of sequence)
length = len(pArray)
# now for plotting. Initialize x and y arrays to plot:
x = []
y = []
# add the average probabilties to the y values:
for i in range(length):
p_i = []
for p in pList:
p_i = p_i + [p[i]]
pAvg = sum(p_i)/len(p_i)
x.append(i)
y.append(pAvg)
# plot the x and y values:
pyplot.plot(x,y)
pyplot.xlabel('position (nt)')
pyplot.ylabel('Averge error probability')
pyplot.savefig('quality.png')
Download the script by running this:wget http://dnacamp.cgrb.oregonstate.edu/untrimmed_reads.fastq
Finally, we can run the script like this:
python ~/scripts/printAverageQualityScores.py untrimmed_reads.fastq
Transfer the file with Filezilla. How does it look?